
第一作者:浙江师范大学博士生刘晓艳,内蒙古大学博士生张铭元
通讯作者:杨启华 教授,刘健 教授,高瑞 教授
通讯单位:浙江师范大学,内蒙古大学
论文DOI:https://doi.org/10.1021/acscatal.4c05501
全文速览
氢的解离和迁移是加氢反应中的关键过程之一,但当反应底物含有硫原子时,这一过程变得极具挑战,甚至不能发生,因为硫原子会严重毒化负载型金属催化剂的活性金属表面。本工作中,我们报道了一种在含硫状态下仍能实现高效氢解离、氢转移与选择性加氢的催化策略,该方法通过氢传递物种与氢气解离位点的协同作用,在Pt/MoO3催化含硫底物5-硝基苯并噻唑加氢反应中得以实现。Pt/MoO3在温和反应条件下表现出99%的转化率和~99%的选择性,是迄今报道的含硫化合物加氢性能最高的催化剂之一。原位-XRD和DFT计算结果表明,Pt/MoO3催化剂的MoO3载体在反应中会储存解离氢,形成的H1.68MoO3中间体具有独特的H溢流和释放性能,不仅加速了5-硝基苯并噻唑中N-O键的断裂以实现加氢,同时还能有效覆盖Pt NPs以防止其被硫毒化。该研究为解决当前含硫化合物催化加氢中的硫毒化挑战提供了一种有前景的策略。
本文亮点:
1. 本工作设计制备的Pt/MoO3催化剂在含硫底物5-硝基苯并噻唑中表现出极高的催化活性,实现了99%的转化率和>99%的选择性。该结果在目前已报道的含硫化合物加氢催化剂中居于最活跃催化剂之列。
2. Pt/MoO3催化剂的H2解离位点和5-硝基苯并噻唑的硝基加氢活性位点分别位于Pt NPs和原位生成的H1.68MoO3,协同促进加氢的同时抗硫中毒。因此,Pt/MoO3具有1440 h⁻¹的高TOF,约为文献报道值的三倍。
3. 机理研究表明活性物种H1.68MoO3在热力学条件下不但易于形成,而且能够将其存储的氢自发释放以还原−NO2基团。反应过程中,H1.68MoO3表面的快速氢转移和固有氧空位是提升其加氢性能的关键因素。
图文解析:
通过共沉淀-空气煅烧法制备的xPt/MoO3催化剂,具有PtOx NPs分布在片状结构的MoO3载体表面(图1a-c),Pt含量分别为0.5,1,2和4 wt%。将其应用在含硫化合物5-硝基苯并噻唑加氢反应中,当反应温度为80 ℃,氢气压力4 MPa,S/C=100(反应底物于Pt摩尔比)时(图d-e),所有金属含量的Pt/MoO3催化剂均具有优异的催化性能,其中2Pt/MoO3催化性能最优异,反应15 min转化率>99%, TOF 为1440 h-1。值得注意的是,当反应条件更温和时(40 ℃,2 MPa H2,S/C=200),2Pt/MoO3依然显示了优异的催化性能,不过其反应初期出现了明显的诱导期(图1f),表明催化剂在反应期间发生了结构演化且生成了活性位点。
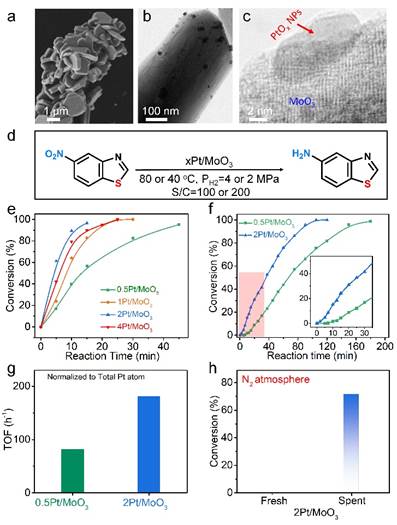
Pt/MoOx催化剂结构和催化性能
对反应后的最优催化剂2Pt/MoO3(命名为spent 2Pt/MoO3)进行表征后发现Pt NPs表面被覆盖(图2a-b),HRTEM,HAADF-STEM,EPR和XPS结果显示该物质为HxMoO3物种(图2c-f)。除此之外,将spent 2Pt/MoO3在氮气气氛下催化5-硝基苯并噻唑进行催化反应12 h(40 ℃,2 MPa N2, S/C=10)后成功得到了71.2%转化率,而新鲜2Pt/MoO3催化剂却不能在氮气气氛下发生催化反应(图1h)。该结果进一步证明在反应过程中原位生成的HxMoO3为活性物种且可以作为H源。
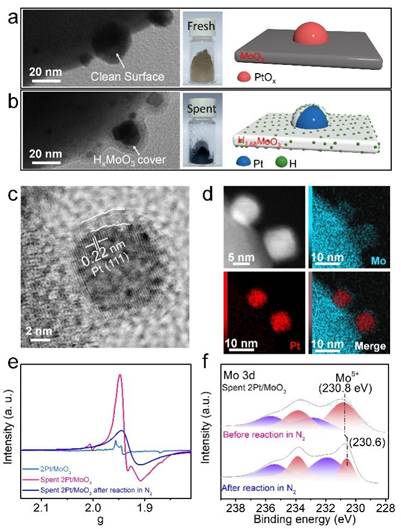
反应后2Pt/MoO3结构表征
原位XRD表征结果显示,Pt/MoO3在氢气气氛下会解离氢并通过氢溢流将活性H物种储存到MoO3体相中形成H1.68MoO3(图3),再次证明原位形成的H1.68MoO3是催化反应发生的活性物种。
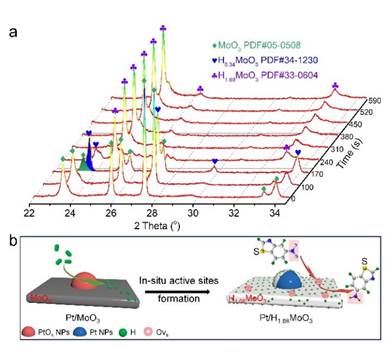
催化剂结构演化表征
为了更深入地解析实验结果,本研究采用密度泛函理论(DFT)对多种HxMoO3物相的稳定性,及其表面反应机理进行了详细研究。首先,通过计算确定了HxMoO3晶体的最稳定结构(图4a),进而计算了其在实验反应温度下的形成吉布斯自由能,结果发现H0.93MoO3为最稳定的物相,与实验数据一致。进一步的计算发现,HxMoO3晶体中的H原子结合能与其相转变之间存在直接关联(图4b),特别是H0.93MoO3与H1.68MoO3之间的可逆性转变。H原子在H1.68MoO3(010)表面上的迁移能垒计算(图4c),进一步证明其快速的氢溢流使得H1.68MoO3成为了一种高效的氢储存库,有助于氢的释放以促进加氢反应。吸附研究进一步显示(图4d),H1.68MoO3(010)表面更倾向于吸附5-硝基苯并噻唑的-NO₂端,有效避免了Pt表面的硫中毒。反应路径分析揭示(图4e),5-硝基苯并噻唑在H1.68MoO3(010)表面的加氢过程涉及N-O键解离形成表面*O和氢迁移脱除*O,并确定了通过H扩散在氧空位上脱除第二个*O原子形成*OH的过程为速率决定步骤,其势垒为0.94 eV。综上所述,由于H1.68MoO3(010)独特的H质子储存和释放性能,不仅促进了5-硝基苯并噻唑氢化过程中N-O键的断裂,而且有效防止了Pt的硫中毒,为开发更高效的催化剂和优化有机化合物氢化反应路径提供了新的见解。
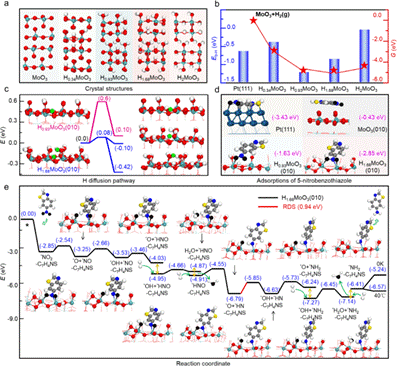
基于DFT研究Pt/MoO3原位演化过程和催化机理
总结与展望:
本工作展示了xPt/MoO3作为一种高效催化剂,可用于含硫化合物5-硝基苯并噻唑(5-nitrobenzothiazole)加氢为5-氨基苯并噻唑(5-aminobenzothiazole)的反应中,该产物是精细化学品和医药的重要中间体,xPt/MoO3表现出优异的抗S毒化性能。我们的实验结果表明,H2和5-硝基苯并噻唑的活化位点分别为Pt和原位形成的H1.68MoO3,这一覆盖在Pt表面的H1.68MoO3物种有效阻止了5-硝基苯并噻唑在Pt上的直接吸附,从而抑制了Pt的硫中毒。DFT计算进一步揭示,H1.68MoO3上自发的氢溢流和可逆相变对于硫化合物加氢至关重要,有助于防止Pt的硫中毒,提升5-硝基苯并噻唑的加氢活性,并作为氢存储和传递的载体。综上所述,实验结果和理论模拟表明H2解离、氢溢流和H1.68MoO3的生成速率是影响xPt/MoO3催化性能的关键因素,这种通过包覆一层薄的H存储和传递层,将H2解离和−NO2加氢活性位点分离开的办法,可能是一种提升金属催化剂在含O、N和S的不饱和化合物加氢反应中活性的有效策略。
